
Introduction
The key diagnostic criteria for Kawasaki disease (KD) are based on clinical symptoms. However, diagnosing the disease based only on these symptoms can be challenging, because several other conditions present similarly, emphasizing the importance of differential diagnosis. In addition to coronavirus disease 2019 (COVID-19)–related multisystem inflammatory syndrome in children (MIS-C) and various viral infections such as adenovirus, rheumatic immune-mediated diseases including systemic juvenile idiopathic arthritis (sJIA) and systemic lupus erythematosus (SLE), as well as macrophage activation syndrome (MAS) associated with these conditions, may also present with clinical manifestations resembling those of KD [1,2]. Previous reports have described cases initially diagnosed as KD that later evolved into MAS or progressed to sJIA [3,4]. Despite these observations, reliable tools to differentiate KD from other systemic inflammatory diseases remain limited.
In this case, the patient presented with fever and hypotensive shock, was confirmed to have influenza infection, and was admitted to the intensive care unit. On the second day of hospitalization, clinical symptoms compatible with KD became apparent, and the patient was treated accordingly and discharged after clinical improvement. However, the patient later returned with fever, generalized joint pain, and limitation of motion, raising suspicion for systemic JIA. The patient was transferred to a tertiary pediatric rheumatology center, where sJIA was diagnosed and treated with subsequent clinical improvement. This case illustrates a sequence involving influenza infection, KD, and sJIA, providing insights into their diagnostic challenges and potential shared immunopathologic mechanisms.
Case
A 5-year-old boy presented to the emergency department with 4 days of fever and gastrointestinal symptoms including abdominal pain, vomiting, and diarrhea. He had previously visited a local clinic where abnormal liver function tests were noted. Initial vital signs were blood pressure 92/55 mmHg, heart rate 147 beats/min, respiratory rate 24 breaths/min, and temperature 39.1 ℃. During observation, blood pressure decreased to 82/35 mmHg. Physical examination showed mild icteric sclera without conjunctival injection, bilateral cervical lymphadenopathy, a truncal maculopapular rash, and right upper quadrant tenderness.
Initial laboratory tests showed hemoglobin 11.7 g/dL, white blood cell count 6,100 /μL (neutrophils 74%, lymphocytes 2%, bands 18%), platelet count 204,000 /μL, erythrocyte sedimentation rate (ESR) 83 mm/hr (normal range 0–9 mm/hr), and C-reactive protein (CRP) 17.98 mg/dL (normal range < 0.5 mg/dL). Additional results included sodium 134.1 mEq/L, albumin 3.1 g/dL, aspartate aminotransferase (AST) 72 U/L, alanine aminotransferase (ALT) 78 U/L, amylase 152 U/L, blood urea nitrogen 51 mg/dL, and creatinine 2.17 mg/dL. NT-proBNP was elevated (4,354 pg/mL), while troponin-I and creatine kinase muscle-brain (CK-MB) were normal. Urinalysis showed pyuria. Abdominal ultrasonography demonstrated borderline hepatosplenomegaly and gallbladder sludge. Initial echocardiography showed preserved cardiac function without coronary artery abnormalities.
Because hypotension with suspected sepsis, acute kidney injury, and hepatopathy was present, the patient was admitted to the intensive care unit. Norepinephrine infusion and empirical antibiotics (vancomycin and meropenem) were started. Influenza A infection was confirmed and oseltamivir was initiated. On hospital day 2, additional clinical findings compatible with complete KD became evident, including cervical lymphadenopathy, polymorphous rash, and mucocutaneous changes. Given the presence of hypotension, Kawasaki disease shock syndrome (KDSS) was suspected, and intravenous immunoglobulin (IVIG) therapy was initiated.
Fever decreased on hospital day 3, although CRP remained elevated. Based on the IVIG resistance scoring system, the patient was considered high risk for IVIG resistance (Table 1) [5]. Methylprednisolone pulse therapy and a second IVIG infusion were administered. Thirty-six hours after the first IVIG infusion, fever recurred and CRP remained elevated (11.18 mg/dL). Because hypotension had been present, KDSS was suspected, and MAS was also considered.
Laboratory tests showed ferritin 662 ng/mL, triglycerides 257 mg/dL, platelet count 166,000 /μL, hemoglobin 10.3 g/dL, AST 22 U/L, and fibrinogen 695 mg/dL. However, these findings did not fulfill the classification criteria for MAS, although early MAS could not be excluded. The patient gradually improved and was treated with oral prednisolone for 15 days. He was discharged on hospital day 9. Seven days after discharge, desquamation of the hands and feet was observed (Fig. 1A). The patient reported hand pain and difficulty fully extending his fingers. There was leukocytosis (28,560 /μL), thrombocytosis (697,000 /μL), CRP 0.09 mg/dL, and ESR 37 mm/hr. Hand radiographs showed no definite abnormalities. These findings were considered consistent with arthritis following severe KDSS.
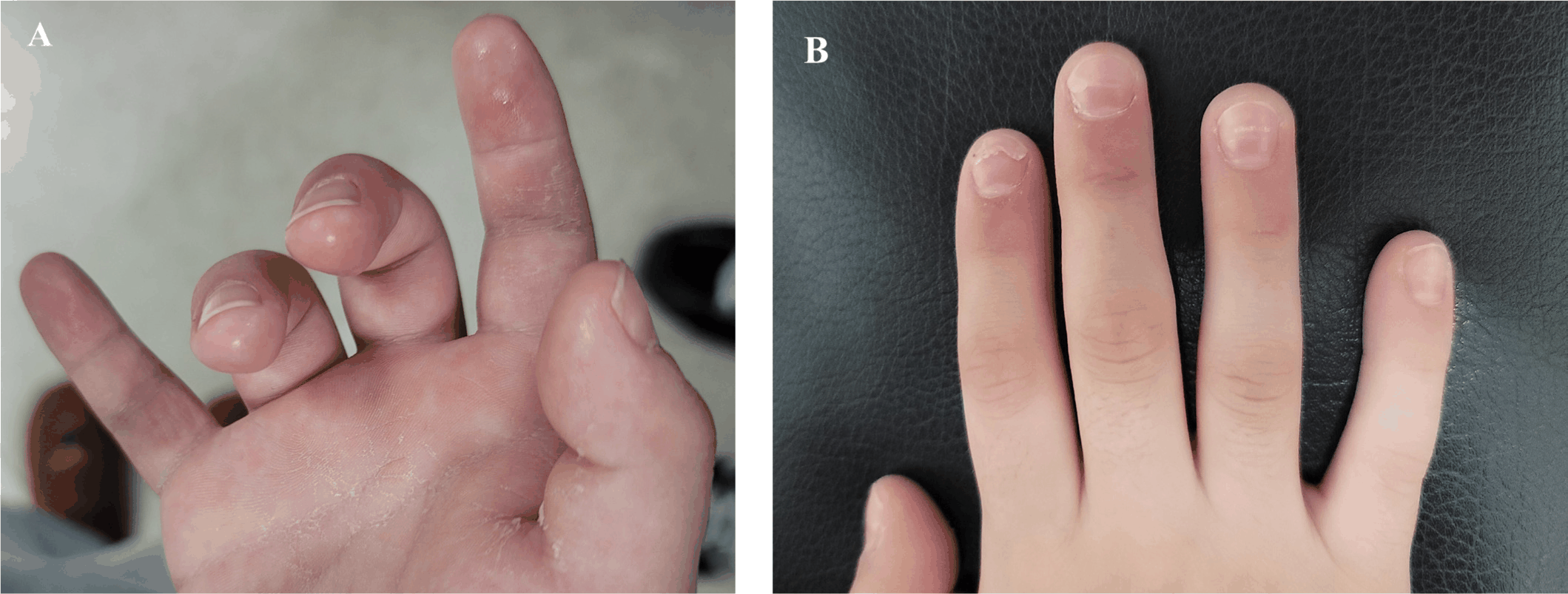
Thirteen days after discharge, the patient returned with recurrent fever and impaired ambulation due to involvement of multiple joints, including the neck, pelvis, and fingers. CRP and ESR were elevated. He was transferred to a tertiary hospital and evaluated for suspected sJIA. Pelvic MRI demonstrated bilateral hip and sacroiliac joint effusion with capsular enhancement of the hip joints. Laboratory studies showed elevated ESR (115 mm/hr), CRP (9.59 mg/dL), and interleukin (IL)-6 (79.7 pg/mL), with negative ANA, RF, anti-CCP, and HLA-B27, and ophthalmologic examination excluded uveitis. Although the strict International League of Associations for Rheumatology (ILAR) classification criteria for sJIA were not yet fully met at the time of diagnosis given the early phase of disease evolution, the overall clinical, laboratory, and imaging findings were considered most consistent with sJIA. Treatment with nonsteroidal anti-inflammatory drugs, weekly methotrexate, and prednisolone was initiated. The symptoms improved, and the patient was discharged on hospital day 9. During subsequent outpatient follow-up, mild joint symptoms persisted but gradually improved. Methotrexate and prednisolone were tapered and discontinued by August 2023, and the patient has remained in sustained clinical remission without immunomodulatory therapy at the most recent follow-up, with no recurrence of fever, arthritis, or systemic inflammation. Follow-up echocardiography performed during the subacute phase after KD treatment showed no coronary artery abnormalities. Nail changes suggestive of Beau’s lines, consistent with the convalescent phase of KD, were observed (Fig. 1B) [6]. The patient continues to undergo regular follow-up in the pediatric rheumatology and pediatric cardiology clinics.
Discussion
This case describes the clinical course of a child initially diagnosed with KDSS in association with influenza infection, who subsequently developed severe arthritis and was managed as sJIA. The clinical course suggests that the early phase represented KDSS followed by sJIA or an evolving systemic inflammatory disease associated with MAS.
MAS was considered during the acute phase because the patient presented with hypotension and systemic inflammation. However, the laboratory findings did not meet the 2016 EULAR/ACR classification criteria for MAS complicating sJIA [7]. Ferritin increased from approximately 400 ng/mL to 662 ng/mL but did not exceed the diagnostic threshold of 684 ng/mL, and other laboratory findings were not fully compatible with MAS. Nevertheless, because the patient was later considered to have sJIA, the early inflammatory phase might have represented a subclinical or evolving MAS-like condition that did not meet diagnostic criteria at the time of evaluation. This highlights the limitation of relying solely on strict diagnostic thresholds during early disease evolution.
Another important aspect of this case is the potential role of viral infection as a trigger. Respiratory viral infections, including influenza, have been reported in association with KD. More recently, interest in the relationship between viral infections and systemic inflammatory syndromes in children has increased following the emergence of MIS-C during the COVID-19 pandemic. Previous studies have suggested that viral infections may trigger abnormal immune activation leading to KD, KD-like illnesses, or systemic inflammatory diseases [8,9]. Although influenza-associated KD has been reported, the clinical features appear to differ from COVID-19-associated KD-like illness. Influenza-associated KD typically occurs in younger children and often involves coronary artery abnormalities, whereas COVID-19-associated inflammatory syndromes tend to occur in older children and more frequently involve myocardial dysfunction [10].
Joint manifestations in KD have also been described. Transient arthritis or arthralgia may occur during the acute phase of KD and is generally self-limited. However, arthritis may also develop during the convalescent phase, typically between 2 and 8 weeks after disease onset. Some patients develop persistent inflammatory arthritis resembling rheumatologic diseases, and differentiation from systemic autoimmune diseases such as sJIA or SLE may be required. Epidemiologic data from Japan suggest that KD-associated arthritis occurs in approximately 48 per 100,000 patients with KD. Arthritis occurring after defervescence has been described as the intermittent fever onset type and may appear during the convalescent phase [11]. A central question was whether the recurrent fever and polyarthralgia after steroid tapering reflected KD recurrence or an evolving rheumatologic disease. The pattern of joint involvement differed from KD-associated arthritis, which is typically oligoarticular and self-limited: our patient developed symmetric polyarthritis of the pelvis, posterior neck, bilateral hips, and small joints of the hands with inability to ambulate, and pelvic MRI demonstrated bilateral hip and sacroiliac joint effusion with capsular enhancement of the hips. Although the strict ILAR criteria for sJIA were not fully met at diagnosis given the early phase of disease, the overall picture—marked systemic inflammation (ESR 115 mm/hr, CRP 9.59 mg/dL, IL-6 79.7 pg/mL), hepatosplenomegaly, negative ANA, RF, anti-CCP, and HLA-B27, exclusion of uveitis and infection, and a sustained response to NSAIDs, methotrexate, and prednisolone rather than to repeat IVIG—was most consistent with sJIA. This diagnostic uncertainty itself reinforces the central message of the report: rigid application of established criteria during early disease evolution may be insufficient, and a shared inflammatory predisposition between KD and sJIA cannot be excluded. In the present case, the patient developed severe polyarticular symptoms approximately two weeks after defervescence, with involvement of multiple joints including the pelvis, neck, knees, and fingers, resulting in significant limitation of motion. The severity and persistence of symptoms, together with elevated inflammatory markers, raised suspicion for a systemic inflammatory disease, leading to a working diagnosis most consistent with sJIA. Although the diagnosis of sJIA was not definitive at that time, the overall clinical picture favored sJIA over other conditions, and treatment was initiated accordingly following a JIA-directed therapeutic approach. Several case reports have also described patients initially diagnosed with KD who were later considered to have sJIA. These observations suggest that overlapping immunopathological mechanisms may exist between KD and systemic inflammatory diseases [11]. Furthermore, MAS may occur in both conditions and can be triggered by infections or medications. In patients with KD, MAS has been associated with more severe inflammation and an increased risk of coronary artery complications [12]. Therefore, careful monitoring and consideration of evolving inflammatory syndromes are important during the clinical course.
Overall, this case highlights the diagnostic complexity encountered when systemic inflammatory diseases present with overlapping clinical features. When patients with KD develop persistent or worsening systemic symptoms, including arthritis, clinicians should consider the possibility of evolving rheumatologic disease or MAS, even if initial laboratory findings do not meet established diagnostic criteria.
In conclusion, this case highlights the diagnostic challenges that may arise when KD presents with viral infection, severe systemic inflammation and subsequent arthritis. The clinical course suggests the possibility of overlapping conditions, including KDSS, MAS, and sJIA. These findings emphasize the importance of continuous clinical reassessment and consideration of evolving inflammatory diseases when patients with KD exhibit atypical or persistent systemic manifestations. Further studies are needed to better understand the shared immunopathological mechanisms among KD, MAS, and systemic inflammatory diseases such as sJIA.