
서론
소아특발관절염(juvenile idiopathic arthritis, JIA)은 소아에서 가장 흔한 류마티스 질환이다[1–3]. 국제류마티스학회(International League of Associations for Rheumatology, ILAR)는 JIA를 전신형(systemic), 소수관절형(oligoarticular), 다수관절형(polyarticular), 건선형(psoriatic), 부착부염 연관형(enthesitis-related), 미분류형(undifferentiated)의 6가지 아형(subtype)으로 분류한다[4]. 이 중 전신형 소아특발관절염(systemic juvenile idiopathic arthritis, SJIA)은 역학적, 임상적, 치료적 측면에서 다른 아형의 JIA와 구분되는 특징을 나타낸다[2,5]. 또한 병태생리 측면에서 소수관절형 JIA나 다수관절형 JIA가 자가면역(autoimmune) 기전에 의해 발생하는 것과 다르게, SJIA는 자가염증(autoinflammatory) 기전과 연관된 것으로 알려져 있다[5–7]. 이러한 차이점을 토대로, 많은 전문가들은 SJIA를 다른 아형의 JIA와 구분해야 하는 별개의 질환으로 간주한다[8–10].
실제 임상에서, SJIA는 관절염을 주 증상으로 하는 근골계 질환보다는 발열과 발진을 주 증상으로 하는 소아기 열성 질환과 혼동되는 경우가 적지 않다[11,12]. 여러 종류의 소아기 열성 질환 중 가와사키병(Kawasaki disease, KD)은 SJIA와 구분해야 하는 가장 중요한 질환들 중 하나이다[13–15]. 따라서, 류마티스 전문가뿐만 아니라 KD 전문가들도 SJIA와 KD의 관계에 대한 이해가 필요하다. 본 연구에서는 SJIA의 개요, SJIA와 KD의 유사점과 차이점, 그리고 두 질환과 관련하여 추가적으로 고려해야 할 사항에 대해 기술하여, KD를 진료하는 임상의에게 SJIA와 KD의 관계를 이해하는 데 도움되는 기본 지식을 제공하려고 한다.
본론
SJIA는 소수관절형(40%–50%), 다수관절형(20%–25%) 다음으로 흔한 JIA의 아형(10%–20%)으로, 발열과 발진, 관절염을 특징으로 한다[3,5]. 호발연령은 1–5세이지만 10세 이후 연장아에서도 발생하며, 16세 이후에 발생할 경우 성인형 스틸병(adult-onset Still's disease, AOSD)이라 부른다[10]. 최근 스틸병(Still’s disease)의 개념이 새롭게 정립되면서, SJIA는 스틸병의 아형 중 하나로 편입되었다[16,17]. 즉, 스틸병을 발병 연령에 따라 i) 소아형(pediatric onset) 스틸병(기존의 SJIA), ii) 성인형 스틸병(기존의 AOSD), iii) 노인형(elderly onset) 스틸병으로 분류한다[18]. 본 연구에서는 소아형 스틸병이라는 용어 대신 16세 미만에서 발병할 때 SJIA로, 16세 이상에서 발병할 때 AOSD로 기존의 용어를 사용하였다.
SJIA의 3대 증상은 발열(~100%)과 발진(> 80%), 관절염(50%–85%)이다[19]. 첫째, 발열은 최소 2주 이상 지속되는데 이는 다른 아형의 JIA와 구분되는 핵심 증상이다. 하루 한두 차례 39 ℃ 이상의 고열(quotidian, spike fever)을 보였다가 빠르게 정상 체온으로 돌아온다(Fig. 1A). 발열이 있을 때는 오한을 동반하며 매우 힘들어 보이지만, 해열이 되면 아파 보이지 않는다[1,19]. 둘째, 발진 양상은 짧은 시간 내 사라지는 반점성 연어 분홍색(evanescent, macular, salmon-pink)이다(Fig. 1B). SJIA의 발진은 진단에 도움되는 특징적 소견이지만, 유사한 반점성 발진이 KD나 다른 류마티스 질환, 홍역, 풍진, 돌발진, 코로나바이러스감염증-19(coronavirus disease 2019, COVID-19) 등 감염 질환, 그리고 아바카비르(abacavir)와 같은 항레트로바이러스제나 페니토인(phenytoin)과 같은 항경련제의 약물 이상반응으로도 발생할 수 있다[20].
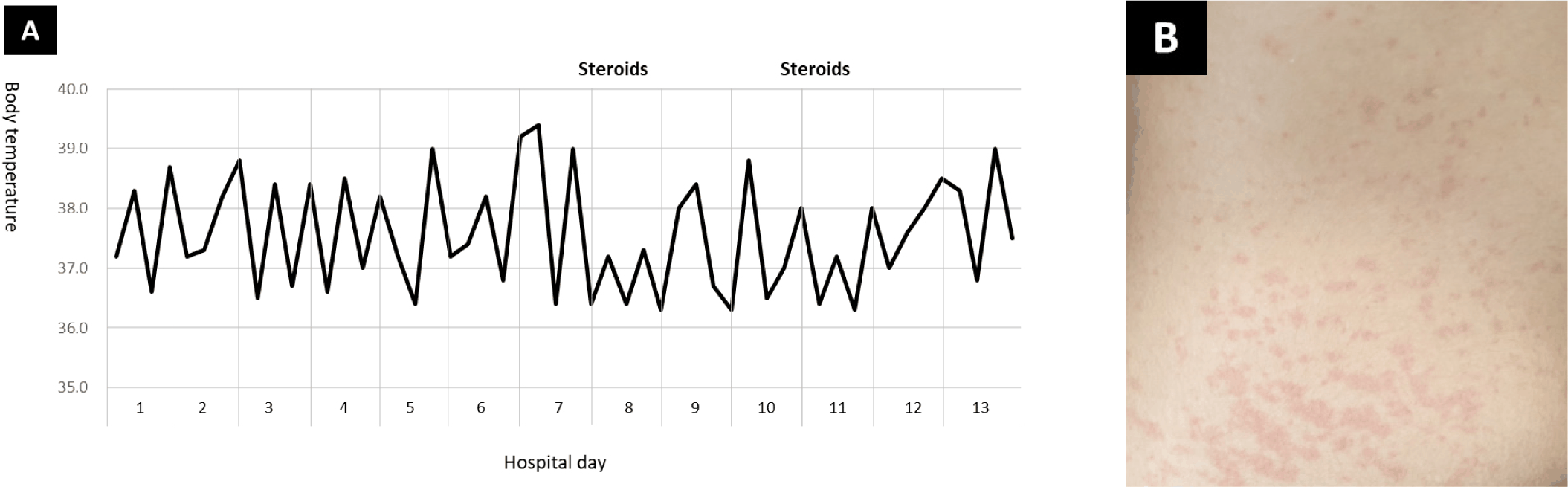
셋째, 관절염은 복수의 관절에서 발생한다. 많은 SJIA 환자들은 질병 초기에 손목, 무릎, 발목에 관절염을 경험한다[16,17]. 하지만 30%–50%의 환자는 질병 후반부에 관절염을 보이며, 상당수의 환자는 관절 증상을 전혀 호소하지 않기도 한다[18]. 따라서 관절 증상이 없더라도, 설명할 수 없는 발열과 발진을 나타내는 소아와 청소년에서 SJIA 가능성을 고려하는 것이 중요하다. 3대 증상 외에, SJIA 환자들은 림프절종대, 간비종대, 인두염을 흔하게 보이며, 심초음파 검사에서 심낭염(pericarditis)이나 관상동맥 이상(coronary artery abnormalities, CAAs)이 확인될 수 있다[21,22]. SJIA의 주요 증상은 ILAR 진단기준에 반영되었다[4]. 흥미롭게도, SJIA의 주요 증상은 KD 환자에서도 관찰된다(Table 1)[23–25].
| SJIA diagnostic criteria [4]1) | Principal features of KD |
|---|---|
| Fever ≥ 2 weeks | Fever ≥ 5 days (diagnostic criteria) |
| Arthritis ≥ 1 joint | Arthritis [23,24] |
| ≥ 1/4 following criteria | |
| Evanescent erythematous rash | Polymorphous rash (diagnostic criteria) |
| Generalized lymphadenopathy | Cervical lymphadenopathy (diagnostic criteria) |
| Hepatomegaly ± splenomegaly | Splenomegaly [25] |
| Serositis: pericarditis, pleuritis, or peritonitis | Pericarditis (common on ECHO) |
To diagnose SJIA, fever + Arthritis + ≥ 1/4 criteria. Arthritis is not an essential criterion in the PRINTO definition [2].
SJIA의 드물지만 심각한 합병증으로 SJIA-연관 폐질환(SJIA-associated lung disease, SJIA/LD)과 대식세포활성증후군(macrophage activation syndrome, MAS)가 발생할 수 있다[26–28]. SJIA/LD에는 폐고혈압(pulmonary hypertension), 간질성폐질환(interstitial lung disease), 폐포단백증(pulmonary alveolar proteinosis), 지질폐렴(lipoid pneumonia)이 포함된다[26]. SJIA/LD는 2013년 이전에는 매우 드문 합병증으로 여겨졌으나, 최근 발생빈도가 증가하는 추세로 SJIA 환자의 5%–7%에서 보고된다[27]. SJIA/LD는 심한 염증반응뿐만 아니라, 생물학제제(biologics) 치료와 연관되어 증가한다고 알려졌다[28].
MAS는 T 세포와 대식세포가 지나치게 활성되어 발생하는 염증 상태로, 심한 전신염증(systemic inflammation)과 장기부전(organ dysfunction)을 특징으로 한다[29–31]. MAS는 SJIA 환자의 ~10%에서 보고된다[29]. MAS는 또한 전신홍반루프스(systemic lupus erythematosus; ~5%)나 KD (~2%)와 같이 심한 염증반응을 유발할 수 있는 다른 소아 질환에서도 발생하기 때문에, SJIA가 진단적으로 혼돈되는 원인이 되기도 한다[30]. SJIA 환자에게 SJIA/LD나 MAS가 합병되면 사망률은 30%–60%로 크게 증가한다[28,31]. 따라서 SJIA 환자에서 이러한 합병증을 조기 인식하기 위하여, 영상 검사와 혈액 검사를 포함한 정기적인 추적관찰을 시행해야 한다.
Table 2에는 SJIA와 KD의 특징을 비교하였다. SJIA와 KD의 호발연령은 5세 이하로 유사하며, 남아의 비율은 KD에서 상대적으로 높다. KD가 우리나라를 포함하여 동아시아 지역에서 호발하는 반면, SJIA는 전 세계적으로 유사한 빈도로 발생한다[3,23]. 두 질환의 병리기전에 감염 유발인자(infectious triggers)가 직접 또는 간접적으로 관여할 것이라 추정되지만, 아직까지 명확히 밝혀진 원인 미생물은 없다[5,9].
| Variables | SJIA | KD |
|---|---|---|
| Demographic/etiology | ||
| Age | 1–5 years & 10–14 years | < 5 years (mean 2.4) |
| Sex | M ≒ F (other JIAs, M < F) | M > F |
| Race/ethnicity | No differences | East Asia |
| Infectious triggers | NA, possible? | Important, but unknown |
| Distinct cytokines | IL-181) | IL-6 |
| Clinical | ||
| Fever | Quotidian, spike ≥ 2 weeks | Persistent ≥ 5 days |
| Rash | Evanescent erythematous | Polymorphous |
| Arthritis | Symmetric (~80%) | Transient (~⅓) |
| Organ dysfunction1) | Possible | Possible (e.g., KDSS) |
| Laboratory | ||
| Neutrophilia | Common | Common |
| Thrombocytosis | Common | Common |
| CRP elevation | Common | Common |
| Anemia1) | Possible2) | Possible2) |
| Abnormal AST/ALT/albumin1) | Possible2) | Possible2) |
| Hyperferritinemia1) | Possible2) | Possible2) |
| D-dimer elevation1) | Possible2) | Possible2) |
| NT-proBNP elevation | Uncommon | Common |
| Treatment/outcomes | ||
| 1st line | DMARDs, steroids | IVIG + aspirin |
| 2nd line | Anakinra, tocilizumab | Steroids, infliximab |
| Cardiac complications |
CAAs (possible), transient? Pericarditis (~40%) |
CAAs (~25%), long-term sequels Pericarditis (common on ECHO) |
| Disease course | Polyphasic, chronic, recurrent | Monophasic, acute, self-limiting |
| Mortality | 0.6% (6/962) [43,44] | 0.02% (2/14,916) [23,42] |
Laboratory findings for severe or atypical forms of SJIA and KD (e.g., active SJIA, refractory KD, or incomplete KD).
SJIA: systemic juvenile idiopathic arthritis; KD: Kawasaki disease; M: male; F: female; NA: not available; IL: interleukin; KDSS: Kawasaki disease shock syndrome; CRP: C-reactive protein; AST: aspartate aminotransferase; ALT: alanine aminotransferase; NT-proBNP: N-terminal pro-brain natriuretic peptide; DMARDs: disease-modifying antirheumatic drugs; IVIG: intravenous immunoglobulin; CAAs: coronary artery abnormalities; ECHO: echocardiogram; SJIA/MAS: SJIA complicated with macrophage activation syndrome; KD/MAS: KD complicated with MAS.
전신염증이 SJIA와 KD의 공통적인 특징이라는 점에서, 두 질환의 병인을 밝히기 위해 다수의 면역학적 연구가 시행되었다[32–35]. 연구 결과에서, SJIA와 KD는 AOSD, PFAPA (periodic fever, aphthous stomatitis, pharyngitis, and adenitis) 증후군, 베체트병(Behçet disease)과 함께, 다유전자성 자가염증 질환군(polygenic autoinflammatory disorders)에 포함된다는 것이 확인되었다[22,32]. 또한, 두 질환의 핵심적인 병리기전은 종양괴사인자-α (tumor necrosis factor-α, TNF-α), 인터루킨-1(interleukin, IL-1), IL-6, IL-18 등 선천면역계(innate immune system) 사이토카인(cytokine)의 질적, 양적 변화라는 것이 밝혀졌다[34,35]. 두 질환은 일부 병리기전을 공유하지만, 면역학적으로 구분되는 특징을 보이기도 한다. 예를 들어 IL-6와 IL-18는 두 질환 모두에서 증가하지만, 실제 농도를 비교하면 IL-6 농도는 SJIA보다 KD에서 상대적으로 높고, 반면에 IL-18 농도는 KD보다 SJIA에서 상대적으로 높다[34].
발열과 발진, 관절통은 SJIA와 KD의 공통적인 증상이다[11,12]. 두 질환의 발열은 항생제 치료에 반응하지 않으며, 다른 소아기 열성 질환보다 연장되는(prolonged) 양상이다. 반점성 연어 분홍색 발진(Fig. 1B)은 SJIA의 진단을 지지해주는 특징적 소견이지만, KD 환자에게도 드물지 않게 관찰된다[15,20]. 관절 증상 역시, 심한 정도와 지속되는 기간에는 차이를 보이지만, KD 환자의 약 1/3에서 확인된다[23,24]. 장기부전은 두 질환의 흔한 증상은 아니지만 심한 형태의 SJIA나 KD 환자에서 관찰되며, 특히 MAS가 합병되었을 때 동반될 수 있다[29,30].
전반적인 검사실 소견은 KD보다 SJIA에서 심하다. 빈혈, 중성구증가증, 혈소판증가증, C-반응단백(C-reactive protein, CRP) 상승, 간효소치 증가, 저알부민혈증, 고페리틴혈증은 활성 SJIA (active SJIA)의 중요한 검사실 소견이다[36,37]. 하지만, 이러한 소견은 치료불응(refractory) KD와 가와사키병 쇼크증후군(Kawasaki disease shock syndrome, KDSS)과 같은 심한 형태의 KD에서도 관찰된다[38–40]. 실제로, CRP 상승, 빈혈, 백혈구증가증, 혈소판증가증, 간효소치 증가, 저알부민혈증은 미국심장학회(American Heart Association, AHA)의 진료지침에서 불완전(incomplete) KD를 진단할 때 활용되는 검사실 지표이다[23].
고페리틴혈증과 D-이합체(D-dimer) 상승은 MAS가 합병되었을 때 동반되는 핵심적인 검사실 소견이다[31]. 따라서 조절되지 않는 발열, 비장종대, 혈구감소증 등, MAS 발생이 임상적으로 의심되는 상황에서, 페리틴과 D-이합체를 포함한 MAS에 대한 선별검사를 시행해야 한다[37,39]. 심장 침범과 연관된 N-말단 뇌나트륨이뇨펩티드(N-terminal pro-brain natriuretic peptide, NT-proBNP)의 상승은 SJIA보다 KD에서 흔하다[41].
두 질환에 대한 기본적인 치료방법은 차이를 보인다(Table 2). 1차 치료제로, KD는 정맥주사 면역글로불린(intravenous immunoglobulin, IVIG)을, SJIA는 질환조절 항류마티스제(disease-modifying antirheumatic drugs, DMARDs)를 사용한다[5,23]. 하지만, 환자의 상태, 감염질환의 동반, 질병의 중등도 등, 임상 상황에 따라 일부 약제들은 두 질환의 치료에 공통적으로 적용된다. 예를 들어, SJIA 환자에 중증 감염질환이 동반되거나 MAS가 합병되었을 때, IVIG를 투여할 수 있고[30], 치료불응 KD 환자에게 DMARDs 또는 면역억제제에 속하는 사이클로스포린(cyclosporine)이나 메토트렉세이트(methotrexate)를 사용하기도 한다[42]. 최근 두 질환에서 생물학제제의 사용 빈도가 증가한다는 것도 중요한 치료적 공통점이다[30,31].
장기간의 비가역적인 심장 합병증은 KD 환자의 치료에서 고려해야 할 가장 중요한 문제이다. 하지만, CAAs와 심막염과 같은 KD의 중요한 심초음파 소견이 SJIA 환자에서 관찰되어 두 질환이 진단적으로 혼돈될 수 있다[21,22]. Go et al.[15]은 SJIA 환자의 CAAs는 모두 Z-score가 2.5 미만이었고 12개월 이내 정상화되었다고 보고하였다. SJIA 환자에서 관찰된 CAAs에 대한 자료가 부족하므로, 이에 대한 추가적 연구가 필요하다.
질병 경과의 만성화 여부는 SJIA와 KD의 가장 중요한 차이점일 것이다[12,15]. SJIA는 수년에 걸쳐 증상의 완화와 악화가 반복되는(polyphasic) 경과를 보인다. KD는 급성기(1–2주), 아급성기(3주), 회복기(6–8주)를 거쳐 2개월 이내 자기 제어적으로(self-limiting) 회복된다[23]. KD 환자의 약 3%는 재발되기도 하지만, 대부분의 환자는 단발성(monophasic) 경과를 보인다. SJIA의 사망률은 생물학제제를 치료에 도입한 후 크게 개선되었으나, 아직까지는 KD보다 높은 것으로 간주된다[23,42–44].
SJIA와 KD의 유사점과 차이점은 두 질환의 감별진단에 적용할 수 있다. Boyarchuk et al.[13]는 i) 상대적으로 심한 임상양상, ii) 고페리틴혈증과 IL-18 증가와 같은 검사실 소견 그리고 iii) 질병 경과의 만성화 여부를 확인함으로써, KD 환자 중에서 SJIA 환자를 선별할 수 있다고 제안하였다.
SJIA와 KD는 독립적인 별개의 질환이지만, 일부 KD 환자는 반복적이고 만성적인 발열과 발진, 관절 증상을 호소하다가 최종적으로 SJIA로 진단되기도 한다[11–15]. 미국 연구[14]에서, 6개월 동안 KD 환자를 추적 관찰하였을 때 환자의 0.2%(10/6,745)는 SJIA로 진행되었다고 보고하였다. SJIA로 진행된 KD 환자들은 그렇지 않은 일반 KD 환자보다 심한 임상 증상(연장된 발열, IVIG 저항성, MAS 합병)과 검사실 소견(백혈구증가증, 저알부민혈증)을 나타냈다. 캐나다 연구[15]에서도, KD 환자의 0.5%(8/1,765)는 최종적으로 SJIA로 진단되었다. SJIA로 진행된 KD 환자 8명 중 5명(62.5%)에서 CAAs가 확인되었는데, 환자들의 모든 병변은 6주 이내 정상화되었다. 최근 대만의 건강보험 자료를 이용한 연구[45]에서도 KD를 경험하지 않은 대조군에 비해, KD 병력이 있던 소아에서 SJIA의 발생 위험도는 2배 이상으로 높았다.
MAS는 두 질환의 공통적인 합병증이다. MAS가 합병된 SJIA(SJIA complicated with MAS, SJIA/MAS)와 MAS가 합병된 KD (KD complicated with MAS, KD/MAS)는 SJIA와 KD의 가장 심한 임상양상으로 간주된다[6,46]. MAS가 합병되면 두 질환의 임상적, 검사실 소견은 좀더 심해지고 유사해진다[7,11]. 또한, 두 질환의 면역학적 소견도 좀 더 유사해진다. 예를 들어, SJIA/MAS 환자와 KD/MAS 환자에서 공통적으로 IL-18과 인터페론-γ (interferon-γ, IFN-γ)의 증가가 확인된다[47–50]. SJIA/MAS와 KD/MAS의 임상적, 면역학적 유사성을 근거로, 일부 전문가들은 KD, SJIA 및 MAS를 하나의 질환 스펙트럼(same disease spectrum)으로 간주할 것을 주장한다[15,50].
MAS는 사망률 30% 이상의 치명적인 합병증으로, 조기에 진단하여 적극적으로 치료하는 것이 중요하다[29]. 페리틴 또는 IL-18의 급증을 확인하여 SJIA 환자 또는 KD 환자에서 MAS 발생 여부를 조기에 인식할 수 있다[47,48]. 또한 일반 SJIA (~10%)와 KD (~2%)보다, 심한 형태의 SJIA (~30%)와 KD (~7%)에서 MAS 발생빈도가 크게 증가하므로[31,39], 적절한 치료에도 불구하고 예상 밖의 심한 임상양상을 보이는 SJIA 환자와 KD 환자에서 MAS 합병 가능성을 고려해야 한다.
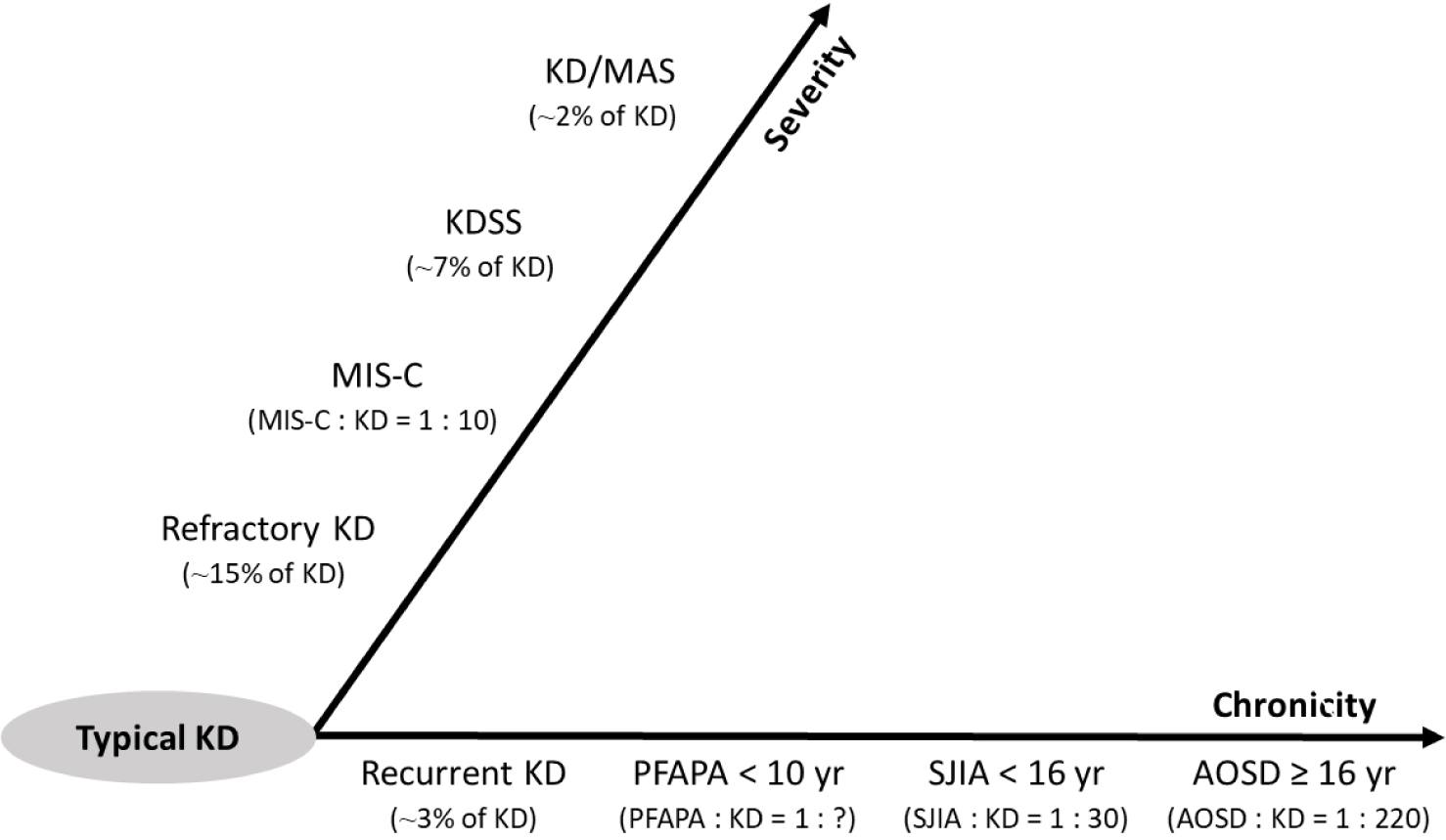
COVID-19 유행 시기에 소아다기관염증증후군(multisystem inflammatory syndrome in children, MIS-C)이 보고되면서, KD-유사증상(KD-like symptoms), 전신염증 및 장기부전을 특징으로 하는 소아 과염증 질환들이 주목받았다[40,50]. ‘KD-유사 과염증 질환(KD-like hyperinflammatory diseases)’에는 KD와 MIS-C뿐만 아니라, 독성쇼크증후군(toxic shock syndrome, TSS), KDSS, KD/MAS, SJIA, SJIA/MAS 등이 포함된다[51].
Fig. 2에는 KD와 KD-유사 과염증 질환들을 중증도(severity)와 만성화(chronicity)에 따라 분류하였다[39,51–55]. 각 질환은 세부적인 병리기전이 다른 독립적인 존재이지만, 전신염증과 장기부전이라는 임상 표현형(clinical phenotype)을 공유한다. SJIA와 KD의 유사점과 차이점을 이해하는 것은 다양한 종류의 소아기 과염증 질환들의 병태생리 연구와 치료전략 개발에 유용한 단서를 제공할 것이다.
결론
SJIA와 KD는 임상양상과 검사실 소견에서 많은 유사점을 보여 진단적으로 혼돈될 수 있다. KD에 비해, SJIA에서 관절 증상과 장기부전이 흔하고 전반적인 검사실 소견도 심하다. 하지만, 특징적인 SJIA의 소견은 심한 형태의 KD에서도 관찰된다는 것을 이해해야 한다. 두 질환의 가장 중요한 차이점은 질병 경과의 만성화 여부일 것이다. 따라서 KD 환자가 발열, 발진 또는 관절 증상을 반복적으로 나타내는 경우, 간과된 SJIA의 가능성을 고려해야 한다. 실제로, KD 환자의 ~0.5%는 최종적으로 SJIA로 진행한다. MIS-C, KDSS와 KD/MAS와 마찬가지로, SJIA를 소아기 KD-유사 과염증 질환에 포함시킬 수 있다.